Discover and read the best of Twitter Threads about #CompChem
Most recents (21)

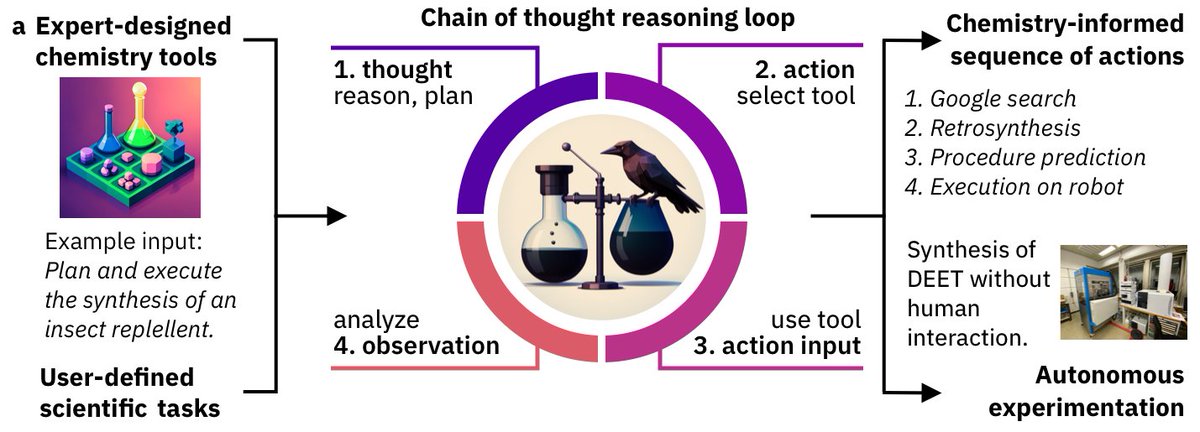
New version of ChemCrow out 🔥🔥 arxiv.org/abs/2304.05376
The LLM-powered chemistry assistant got major updates 💪 What's new?
Robots synthesizing stuff, human/crow collaboration, novel molecules, safety, new evaluations + open source release! 🤩
See more 👇 1/8
#compchem
The LLM-powered chemistry assistant got major updates 💪 What's new?
Robots synthesizing stuff, human/crow collaboration, novel molecules, safety, new evaluations + open source release! 🤩
See more 👇 1/8
#compchem
ChemCrow+chemists collaborate to discover a novel chromophore!
ChemCrow cleans data, train ML model, propose novel molecule with target property + synth procedure. Synthesis yields a product with a wavelength within the model's error!
#Crowlaboration is key 💪
2/8
ChemCrow cleans data, train ML model, propose novel molecule with target property + synth procedure. Synthesis yields a product with a wavelength within the model's error!
#Crowlaboration is key 💪
2/8

Can it be independent tho? YES
ChemCrow + RoboRXN @ForRxn synthesized an insect repellent + 3 organocatalysts, ✨by itself✨
Simple as "synthesize a molecule that's useful for this task". Proof of concept, but imagine a future with functional molecules on demand! 🤩🤩
3/8
ChemCrow + RoboRXN @ForRxn synthesized an insect repellent + 3 organocatalysts, ✨by itself✨
Simple as "synthesize a molecule that's useful for this task". Proof of concept, but imagine a future with functional molecules on demand! 🤩🤩
3/8


🚨Deep learning for large-scale biomolecular dynamics is here!🚨
Today, our group is releasing new work showing how the SOTA accuracy of Allegro can be scaled to massive biomolecular systems up to the full HIV capsid at 44 million atoms!
arxiv.org/pdf/2304.10061… #compchem
1/🧵
Today, our group is releasing new work showing how the SOTA accuracy of Allegro can be scaled to massive biomolecular systems up to the full HIV capsid at 44 million atoms!
arxiv.org/pdf/2304.10061… #compchem
1/🧵
We scale a large, pretrained Allegro model on various systems, from DHFR at 23k atoms, to Factor IX at 91k, Cellulose at 400k, the all-atom fully solvated HIV capsid at 44 million all the way up to >100 million atoms. 2/

This is all done with a pretrained Allegro model w/ 8 million weights at high accuracy of a force error of 26 meV/A, trained on 1mn structures at hybrid functional accuracy using the amazing SPICE dataset. At 8 million weights this is a large+powerful model were scaling here. 3/

(1/5) The latest from our group: Jenna Fromer's overview of computer-aided multi-objective optimization in small molecule discovery is now online & open access @Patterns_CP | doi.org/10.1016/j.patt… #compchem

(2/5) Our focus here is on Pareto optimization. Pareto optimization introduces additional algorithmic complexities, but reveals more information about the trade-offs between objectives and is more robust than scalarization approaches
(3/5) We highlight the extensions from single-objective Bayesian optimization to multi-objective Bayesian optimization when choosing molecules from a discrete library. The primary difference is the definition of the acquisition function, with a few options listed in the fig above


Our team working on ML for chemistry/drug discovery was unfortunately affected by the recent layoffs at Google.
I’m still very much interested in how new technologies can accelerate research in the life and natural sciences.
#ml #ai #compchem #drugdiscovery #googlelayoffs
I’m still very much interested in how new technologies can accelerate research in the life and natural sciences.
#ml #ai #compchem #drugdiscovery #googlelayoffs
If you know of open roles for which expertise in ML, simulation, or modelling as applied to drug/protein/materials design is sought, please feel free to reach out -- any pointer will be much appreciated!
As an opportunity for change, I’m also open to roles that may stretch my skills and expertise. Due to personal constraints, I’ll be looking for positions primarily in the SF Bay and Boston areas.

Fresh!!
Fully differentiable Hückel model using #JAX.
i) We optimize the atom types for different molecular frameworks to find the molecule with the targeted property (HOMO-LUMO) 🧵 (1/3)
@kjelljorner @robpollice @A_Aspuru_Guzik @chemuoft
#compchem
Fully differentiable Hückel model using #JAX.
i) We optimize the atom types for different molecular frameworks to find the molecule with the targeted property (HOMO-LUMO) 🧵 (1/3)
@kjelljorner @robpollice @A_Aspuru_Guzik @chemuoft
#compchem
ii) Parameter optimization a la machine learning for diff. molecular property, for example, polarizability
30% improvement with only 100 DFT training data.
🧵 (2/3)
30% improvement with only 100 DFT training data.
🧵 (2/3)

#arxiv arxiv.org/abs/2211.16763 #github
(inverse molecular design)
github.com/RodrigoAVargas…
(parameter optimization)
github.com/RodrigoAVargas…
🧵 (3/3)
(inverse molecular design)
github.com/RodrigoAVargas…
(parameter optimization)
github.com/RodrigoAVargas…
🧵 (3/3)

🚨Happy to share our newest labor of (nerd) love
🤓❤️
In situ dynamics of influenza glycoproteins illuminate their vulnerabilities
w/ @kanekiyom @LCasalino88 @chem_christian, J Lederhofer, Y Tsybovsky, I Wilson
#glycotime #compchem
biorxiv.org/content/10.110…
🧵:
🤓❤️
In situ dynamics of influenza glycoproteins illuminate their vulnerabilities
w/ @kanekiyom @LCasalino88 @chem_christian, J Lederhofer, Y Tsybovsky, I Wilson
#glycotime #compchem
biorxiv.org/content/10.110…
🧵:
We characterize dynamics of flu glycoproteins in a crowded protein environment via mesoscale (120 nm diameter, 160 million atom) molecular dynamics simulations of 2 evolutionary-linked glycosylated influenza A whole-virion models

Our simulations indicate that the interactions of proteins & glycans in the crowded viral surface facilitate 3 major glycoprotein motions, each of which reveals antigenically relevant states
First, we see an extensive tilting of the neuraminidase (NA) head...
First, we see an extensive tilting of the neuraminidase (NA) head...

Thrilled to share our preprint on structure-based discovery and optimization of SARS-CoV-2 macrodomain (Mac1) inhibitors. We present several low- to sub-micromolar inhibitors and atomic SAR thanks to >150 crystal structures!
biorxiv.org/content/10.110…
biorxiv.org/content/10.110…
Super proud and grateful to be part of an amazing team including scientists from the @BShoichet @fraser_lab @IvanAhelLab @Ashworth_SF @GestwickiLab @adamrenslo @chem4biology and @FrankvonDelft labs!
BIG shoutout to Galen (@fraser_lab) for soaking roughly 300 molecules and determining many many many many crystal structures at 1 Å resolution. All of which are about to be released in the PDB @buildmodels and fragalysis.diamond.ac.uk (Thanks @Darenfearon)

Delighted to introduce FEgrow: An Open-Source Molecular Builder and Free Energy Preparation Workflow! By @bieniekmat @ChemCree @rachaelpirie203 @joshhorton93 @drnataliej
Now out on @ChemRxiv #compchem: doi.org/10.26434/chemr…
🧵tweetorial...
Now out on @ChemRxiv #compchem: doi.org/10.26434/chemr…
🧵tweetorial...
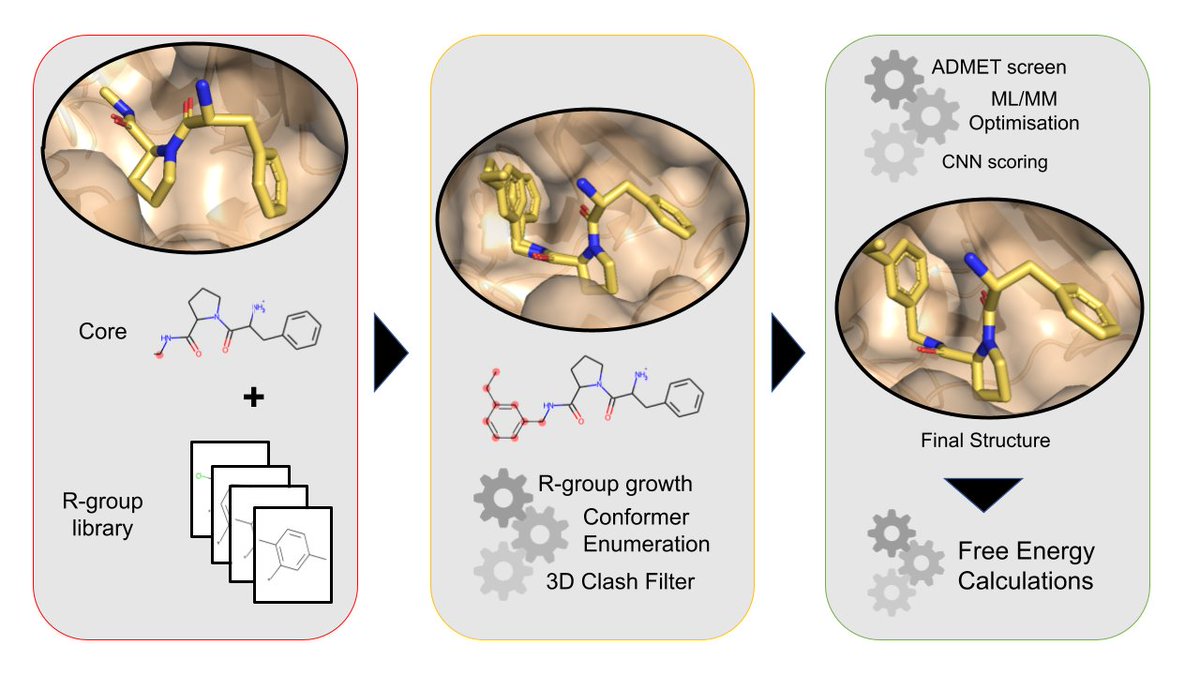
Let's say we have the structure of a hit compound and want to build a congeneric series of ligands bound to a protein. A common task in free energy setup.
For our example, we'll take structures from this very nice study by @JorgensenWL & coworkers: doi.org/10.1021/acscen…
For our example, we'll take structures from this very nice study by @JorgensenWL & coworkers: doi.org/10.1021/acscen…
We can use the very cool mols2grid package to interactively select functional groups to add to the core from a library.

🚨 What comes after Neural Message Passing? 🚨
Introducing Allegro - a new approach:
- no message passing or attention
- new SOTA on QM9+MD17
- scales to >100 million atoms
- 1 layer beats all MPNNs+Transformers
- blazing fast
- theory
arxiv.org/abs/2204.05249
How?👇#compchem
Introducing Allegro - a new approach:
- no message passing or attention
- new SOTA on QM9+MD17
- scales to >100 million atoms
- 1 layer beats all MPNNs+Transformers
- blazing fast
- theory
arxiv.org/abs/2204.05249
How?👇#compchem

First and foremost: this was joint with my co-first author and good friend Albert Musaelian with equal first-author contribution as well as with lab members Anders Johansson, Lixin Sun, Cameron Owen + Mordechai Kornbluth and of course @BKoz / Boris Kozinsky
Message Passing Neural Networks have taken molecular ML by storm and over the past few years, a lot of progress in Machine Learning for molecules and materials has been variations on this theme.
🚀🚨 Equivariance changes the Scaling Laws of Interatomic Potentials 🚨🚀
We have updated the NequIP paper with up to 3x lower errors + (still) SOTA on MD17
Plus: equivariance changes the power-law exponent of the learning curve!
arxiv.org/abs/2101.03164…
👇🧵 #compchem #GNN
We have updated the NequIP paper with up to 3x lower errors + (still) SOTA on MD17
Plus: equivariance changes the power-law exponent of the learning curve!
arxiv.org/abs/2101.03164…
👇🧵 #compchem #GNN

Learning curves of error vs training set size typically follow a power law: error = a * N^b, where N is the number of training samples and the exponent b determines how fast a method learns as new data become available.

Interestingly, it has been found that different models on the same data set usually only shift the learning curve, but do not change the power-law exponent, see e.g. [1]
[1] arxiv.org/abs/1712.00409
[1] arxiv.org/abs/1712.00409

Our @ChemicalScience paper on the discovery of peptide inhibitors of SARS-CoV-2 Mpro is now out: pubs.rsc.org/en/content/art… #CompChem 1/n
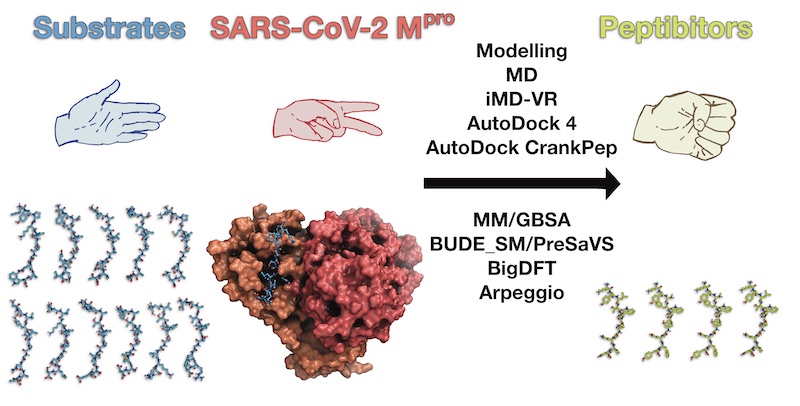
Thank you to all of my brilliant collaborators—some of whom who are on Twitter: @MarcMoesser (one of the @opiglets) @henryhtchan @TikaRMalla @beccawalters95 @Tristan81946826 @AdrianMulholla1 @alessio_lodola @vicente_moliner @SchofieldOxford @fjduarteg
2/n
2/n

All relevant models, data, input files and code are freely available on GitHub: github.com/gmm/SARS-CoV-2… 3/n

I gave our annual State of the Lab address this week! 🚀
I shamelessly borrowed the idea from Ken Dill--an incredibly clear communicator--who would annually dive into what we had done, how we got there, and where we were going.
Read on if you're interested in the gory details.
I shamelessly borrowed the idea from Ken Dill--an incredibly clear communicator--who would annually dive into what we had done, how we got there, and where we were going.
Read on if you're interested in the gory details.

What is our shared goal, as a lab?
We aim to transform drug discovery from a research enterprise into an engineering science.
Industrializing the production of new medicines is essential to enable us to have a significant positive impact on human health and disease.
We aim to transform drug discovery from a research enterprise into an engineering science.
Industrializing the production of new medicines is essential to enable us to have a significant positive impact on human health and disease.

It's well known that drug discovery has a high failure rate.
Circumstances may have improved a bit since this iconic depiction of the "failure cascade", but there's no debating that the overall success rate is low and costs are incredibly high.
nature.com/articles/nrd30…
Circumstances may have improved a bit since this iconic depiction of the "failure cascade", but there's no debating that the overall success rate is low and costs are incredibly high.
nature.com/articles/nrd30…

We're excited to introduce NequIP, an equivariant Machine Learning Interatomic Potential that not only obtains SOTA on MD-17, but also outperforms existing potentials with up to 1000x fewer data! w/ @tesssmidt @Materials_Intel @bkoz37 #compchem👇🧵 1/N
arxiv.org/pdf/2101.03164…
arxiv.org/pdf/2101.03164…

NequIP (short for Neural Equivariant Interatomic Potentials) extends Graph Neural Network Interatomic Potentials that use invariant convolutions over scalar feature vectors to instead utilize rotation-equivariant convolutions over tensor features (i.e. scalars, vectors, ...). 2/N
We benchmark NequIP on a wide variety of molecules+materials: we start with atomic forces from MD-17 with 1,000 training configurations and find that we not only outperform other deep neural networks, but also perform better or sometimes on par with kernel-based methods. 3/N


Twitter loves to bash B3LYP, so let's put that energy to good use. Any suggestions of a functional for TM thermochemistry/kinetics that is:
1. In ORCA/Gaussian (w/ support for opts & analytical freqs!)
2. Not a pain to converge the SCF
3. Decent for spin states, ideally
#compchem
1. In ORCA/Gaussian (w/ support for opts & analytical freqs!)
2. Not a pain to converge the SCF
3. Decent for spin states, ideally
#compchem
Currently, I feel like these requirements leave me with ωB97X-D as promising, although I know there are likely better options (e.g. ωB97M-V or ωB97X-V) if I didn't have the above constraints (especially #1). MN15 could be promising, but so many parameters make me a bit queasy...
I should mention that I have also posted this on @StackMatter (mattermodeling.stackexchange.com/questions/2473…), but I trust you Twitter folks more. Feel free to answer there if you have an account, and I'll upvote you.

Taking chemical reaction prediction models one step further in a great collaboration with Giorgio (@Giorgio_P_), a brilliant organic chemist!
A thread ⬇️1/N
A thread ⬇️1/N
A major limitation of current deep learning reaction prediction models is stereochemistry. It is not taken into account by graph-neural networks and a weakness of text-based prediction models, like the Molecular Transformer (doi.org/10.1021/acscen…).
How can we improve? 2/N
How can we improve? 2/N
In this work, we take carbohydrate reactions as an example. Compared to the reactions in patents (avg. 0.4 stereocentres in product), carbohydrate contain multiple stereocentres (avg. >6 in our test set), which make reactivity predictions challenging even for human experts. 3/N


Honest question: how many of the rushed papers for #COVID19 drug predictions have led to experimental testing and subsequent promise? I've heard the "reason" that docking-only and AI-only papers are being pumped out is because we need solutions to the pandemic, and fast. 1/n
It's now been 3-4 months since a lot of that work started being published, and where has it got anyone? Have wet-labs picked up the ongoing stories of 1M, 100M, 600M docked molecules and tested any of them? How about the "AI discovers potential inhibitors of this or that"? 2/n
I'd be happy to be convinced that all this work was put to good use by someone, but somehow I'm doubtful. Certainly not in substantial amounts? Ultimately, in the 3-4 months that have passed, if more complete work was done, we'd have promising results now, no? So why rush? 3/n
#compchem folks: best programs for visually editing CIFs? Primary need is for adding molecules to a structure defined in a CIF (but also deleting/moving atoms, rotating atom groups, etc.). I am very dissatisfied with nearly every program I've tried the past several years. Ideas?
- VESTA is great for visualizing, and that's it.
- ASE is fine but really not very practical with regards to the GUI.
- @SAMSONConnect seems super promising but can't write CIFs currently.
- CrystalMaker is good for viewing, not editing.
- Materials Studio is way too pricey
- ASE is fine but really not very practical with regards to the GUI.
- @SAMSONConnect seems super promising but can't write CIFs currently.
- CrystalMaker is good for viewing, not editing.
- Materials Studio is way too pricey
There are tons of excellent tools for non-periodic XYZ files, but for CIFs even just adding a bunch of molecules is a huge pain. Please tell me I'm missing something!
Looking for a weekend/holiday read?
Happy to share this major update of our #NeurIPS2019 #ML4PS workshop paper on chemical reaction classificaction (but not only.. 🧪⚗️🌍). @IBMResearch @unibern #compchem #RealTimeChem
Summary thread ⬇️:
Happy to share this major update of our #NeurIPS2019 #ML4PS workshop paper on chemical reaction classificaction (but not only.. 🧪⚗️🌍). @IBMResearch @unibern #compchem #RealTimeChem
Summary thread ⬇️:
We compared different RXN classification methods. 📍Using a BERT model borrowed from NLP, we matched the ground truth (Pistachio, @nmsoftware) with an accuracy of 98.2%.

We did not only visualize what was important for the class predictions by looking at the different attention weights...

Check out our work on Data-Driven Chemical Reaction Classification at the ML for Physical Sciences poster session (15.20-16.20, room 109) #NeurIPS2019 #compchem @IBMResearch

@IBMResearch @acvaucher @teodorolaino The reaction map was visualized using @skepteis's TMAP library (-> tmap.gdb.tools) @reymondgroup
Our Molecular Transformer paper has been accepted to the ML for Molecules and Materials workshop at #NIPS2018. Looking forward!
Paper: bit.ly/2APtF8I
Platform: rxn.res.ibm.com
@IBMResearch @Cambridge_Uni
#RealTimeChem #Chem4Life #CompChem #RXNforChemistry
Paper: bit.ly/2APtF8I
Platform: rxn.res.ibm.com
@IBMResearch @Cambridge_Uni
#RealTimeChem #Chem4Life #CompChem #RXNforChemistry
Our chemical reaction prediction model was built using OpenNMT @harvardnlp and the #NMT transformer architecture by @ashVaswani et al..
Most of the work would not have been possible without @RDKit_org, @dr_greg_landrum, @nmsoftware and @dan2097. Many thanks!
A treasure trove for #nanotechnology experts. Our high-throughput study on novel #2dmaterials. #graphene-like #materials #compchem #dft
Paper: rdcu.be/Gpxi (1/3)
Paper: rdcu.be/Gpxi (1/3)
All the computed data including the full AiiDA #provenance is available on #MaterialsCloud: archive.materialscloud.org/2017.0008/v1 @AiiDA_team (2/3)
Or in a more traditional way in the supplementary information - I would call it the book of 2d materials, definitely worth to have a look at it! static-content.springer.com/esm/art%3A10.1… #BookOf2D (3/3)

